Help
Below are some commonly asked questions which may be able to help solve your problem. If you're still having troubles please contact us on contact@ccs-ties.org.
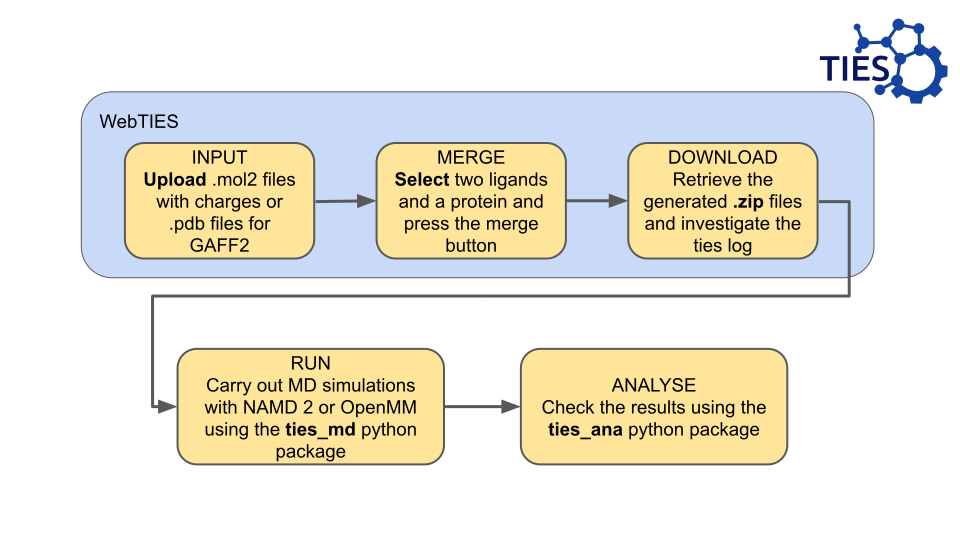
At the moment we encourage using .mol2 files with charges assigned.
Whereas .pdb files will work, they require AM1-BCC charges to be assigned which is computationally expensive for us.
You can upload as many ligand files as you want.
However, at the moment, you will only be able to run two ligand files through TIES at a time. In the future we are planning to increase functionality to allow for more ligands to be analysed at once.
For more information about the TIES software you can read our recently published the paper "TIES 20: Relative Binding Free Energy with a Flexible Superimposition Algorithm and Partial Ring Morphing" in the JCTC.
Feel free to contact us if you have a specific question, and please note that we are hoping to expand our WebTIES service.
The software is free to use for anyone.
However, we reserve the right to access files if an error in the software occurs, allowing us to improve our service.
We are not intending to delete any data until the space becomes an issue. When that happens, we will issue a 2 week warning and select a cutoff date for any old files to be deleted.
Yes, removing the ligand completely deletes the file and all associated metadata.
The initial WebTIES release can fail for different reasons. The generated failed zip will contain the log (ties.log) as well as all files that can help in debugging the issue. We are happy to assist you at contact@ccs-ties.org.
For a higher throughput, yes. However, a small set of GPUs can perform very well. We are currently not making our resources available for the computationally heavy part of running molecular dynamics simulations.
Change Log
Version 1.1 released on Nov 10, 2022
- The default ligand FF is now udpated to use GAFF2 instead of GAFF.
Version 1
- Release